Por Drª Patrícia Pontes | Médica Geneticista – UFBA Fellowship em Neurogenética e EIM – HC-FMUSP – Fellowship DASA Genômica
A doença de Charcot-Marie-Tooth (CMT) é um grupo heterogêneo de neuropatias periféricas hereditárias caracterizadas por degeneração progressiva dos nervos periféricos. Há variabilidade da progressão e gravidade dos sintomas entre os indivíduos afetados.
A Doença de Charcot-Marie-Tooth (CMT) é um grupo de neuropatias hereditárias caracterizadas por neuropatia sensitivo-motora. Sua prevalência global é estimada em 40 casos por 100.000 habitantes. CMT é causada por variantes patogênicas em genes associados à mielina, junções gap e estruturas axonais dos nervos periféricos. Em 1989, a duplicação do gene PMP-22 foi descoberta por Vance et al., e mais de 100 genes foram associados à CMT desde então. Na região Sul do Brasil, variantes dos genes PMP22 e GJB1 são frequentes, representando 38,18% e 9,09% dos casos de uma coorte brasileira.
A CMT é classificada em tipos específicos, incluindo CMT1, CMT2, CMT3, CMT4 e CMTX, cada um subcategorizado por subtipos com letras:
• CMT1: Representa mais de 50% dos casos, com a duplicação do gene PMP-22 causando neuropatia desmielinizante e redução na velocidade de condução nervosa.
• CMT2: Caracteriza-se por neuropatia axonal, com a degeneração dos axônios. O gene mitocondrial MFN2 é um exemplo.
• CMT3: Envolve neuropatias periféricas graves e de início precoce devido à produção inadequada de mielina pelas células de Schwann, resultando em bainhas de mielina finas. Exemplos incluem a síndrome de Dejerine-Sottas e neuropatia hipomielinizantecongênita.
• CMT4: Inclui formas recessivas como a CMT4A, causada por variantes no gene GDAP1. Caracteriza-se por neuropatia sensorial motora desmielinizante com início precoce e progressão variável.
• CMTX: Causada por variantes no gene GJB1, que codifica a conexina 32, essencial para a mielina. Herdada de forma ligada ao X, pode apresentar sintomas semelhantes a outras formas de CMT.
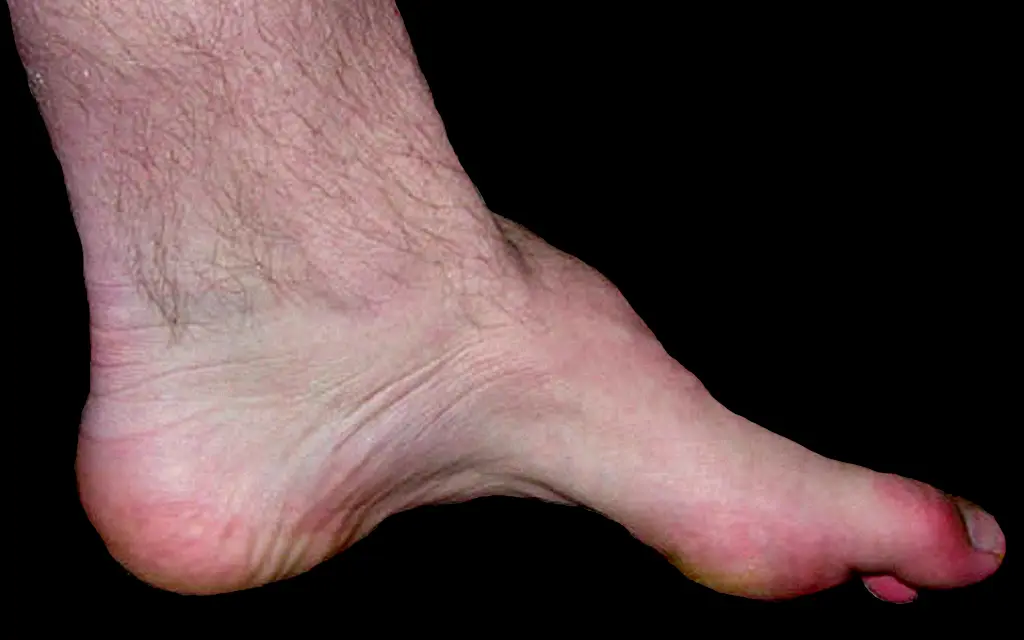
Os sintomas geralmente incluem fraqueza e atrofia muscular de predomínio distal e alterações na sensibilidade, como hipoestesia e parestesias. Deformidades ortopédicas, como pé cavo e dedo em martelo, também são comuns. O diagnóstico envolve avaliação clínica, eletroneuromiografia (ENMG) para diferenciar entre neuropatia desmielinizante e axonal, e testes genéticos para confirmar o tipo de CMT. Para diagnóstico diferencial podem ser solicitado biópsias de nervo e ressonâncias magnéticas são opções mais raras para serem solicitadas, auxilia para diagnóstidiferencial. O tratamento é multiprofissional, incluindo controle da dor, fisioterapia, uso de órteses e dispositivos de assistência, e, se necessário, correção cirúrgica para deformidades. O diagnóstico precoce e a intervenção adequada são cruciais para melhorar a qualidade de vida dos pacientes.
Associação brasileira CMT: https://abcmt.org.br
Associação americana CMT: https://www.cmtausa.org
Referências
• Rossor AM, Polke JM, Houlden H, Reilly MM. Clinical implications of genetic advances in Charcot-Marie-Tooth disease. Nat Rev Neurol. 2013 Oct;9(10):562-71. doi: 10.1038/nrneurol.2013.179. Epub 2013 Sep 10. PMID: 24018473.
• Cavalcanti EBU, Leal RCC, Marques Junior W, Nascimento OJMD. Charcot-Marie-Tooth disease: from historical landmarks in Brazil to current care perspectives. ArqNeuropsiquiatr. 2023 Oct;81(10):913-921. doi: 10.1055/s-0043-1770348. Epub 2023 Aug 23. PMID: 37611635; PMCID: PMC10631856.
• Bird TD. Charcot-Marie-Tooth Hereditary Neuropathy Overview. 1998 Sep 28 [Updated 2024 Aug 1]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1358/
• Yiu EM, Bray P, Baets J, Baker SK, Barisic N, de Valle K, Estilow T, Farrar MA, FinkelRS, Haberlová J, Kennedy RA, Moroni I, Nicholson GA, Ramchandren S, Reilly MM, Rose K, Shy ME, Siskind CE, Yum SW, Menezes MP, Ryan MM, Burns J. Clinical practice guideline for the management of paediatric Charcot-Marie-Tooth disease. J Neurol Neurosurg Psychiatry. 2022 May;93(5):530-538. doi: 10.1136/jnnp-2021-328483. Epub 2022 Feb 9. PMID: 35140138.
• Higuchi Y, Takashima H. Clinical genetics of Charcot-Marie-Tooth disease. J Hum Genet. 2023 Mar;68(3):199-214. doi: 10.1038/s10038-022-01031-2. Epub 2022 Mar 18. PMID: 35304567.

